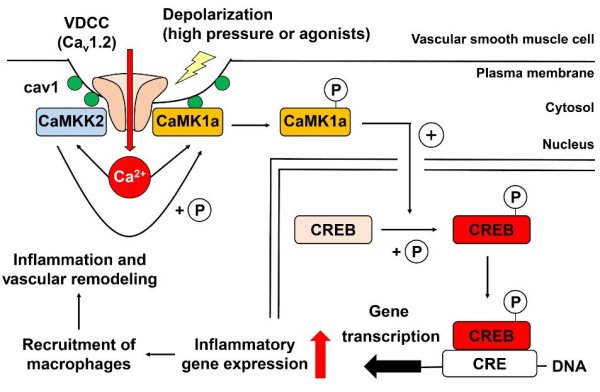
Elevation of intracellular Ca2+ concentration ([Ca2+]i) activates Ca2+/calmodulin-dependent kinases (CaMK) and promotes gene transcription. This signaling pathway is referred to as excitation–transcription (E-T) coupling. Although vascular myocytes can exhibit E-T coupling, the molecular mechanisms and physiological/pathological roles are unknown. Multiscale analysis spanning from single molecules to whole organisms has revealed essential steps in mouse vascular myocyte E-T coupling. Upon a depolarizing stimulus, Ca2+ influx through Cav1.2 voltage-dependent Ca2+ channels activates CaMKK2 and CaMK1a, resulting in intranuclear CREB phosphorylation. Within caveolae, the formation of a molecular complex of Cav1.2/CaMKK2/CaMK1a is promoted in vascular myocytes. Live imaging using a genetically encoded Ca2+ indicator revealed direct activation of CaMKK2 by Ca2+ influx through Cav1.2 localized to caveolae. CaMK1a is phosphorylated by CaMKK2 at caveolae and translocated to the nucleus upon membrane depolarization. In addition, sustained depolarization of a mesenteric artery preparation induced genes related to chemotaxis, leukocyte adhesion, and inflammation, and these
changes were reversed by inhibitors of Cav1.2, CaMKK2, and CaMK, or disruption of caveolae. In the context of pathophysiology, when the mesenteric artery was loaded by high pressure in vivo, we observed CREB phosphorylation in myocytes, macrophage accumulation at adventitia, and an increase in thickness and cross-sectional area of the tunica media. These changes were reduced in caveolin1-knockout mice or in mice treated with the CaMKK2 inhibitor STO609. In summary, E-T coupling depends on Cav1.2/CaMKK2/CaMK1a localized to caveolae, and this complex converts [Ca2+]i changes into gene transcription.
This ultimately leads to macrophage accumulation and media remodeling for adaptation to increased circumferential stretch.